-

-
-

-
-

-
-

- 010-56548231
-

![]()

![]()

![]()

![]()
![]()
Molpro 是一个用于高级分子电子结构计算的从头算程序的综合系统,由 H.-J. Werner 和 P. J. Knowles设计和维护,并包含许多其他作者的贡献。 它包括用于标准计算化学应用的高效且并行化良好的程序,例如具有大量泛函选择的 DFT,以及最先进的高级耦合簇和多参考波函数方法。电子激发态可以使用 MCSCF/CASSCF、CASPT2、MRCI 或 FCI 方法,或通过 TDDFT、CC2 和 EOM-CCSD 等响应方法来处理。有许多模块用于计算分子特性、几何优化、谐波和非谐波振动频率的计算以及进一步的波函数分析。分析能量梯度可用于 DFT、HF、MP2、MP2-F12、CCSD、CCSD-F12、DCSD、QCISD、QCISD(T)、CASSCF 和 CASPT2。密度拟合(DF 或 RI)近似可以将大基组的 DFT 和 MP2 计算速度提高几个数量级,和明确相关的方法 [MP2-F12, CCSD(T)-F12, CASPT2-F12, MRCI-F12] 最小化基组不完整性误差,以产生接近 CBS 质量的三 zeta 基组结果。结合局部近似和高效并行,高级方法 [PNO-LMP2-F12, PNO-LCCSD(T)-F12] 可以应用于化学相关的大分子,产生前所未有的准确性(最近的评论见电线计算机分子科学。2018,e1371)。此外,WF-in-DFT 嵌入或 QM/MM 方法可用于将ab initio方法的适用性扩展到具有化学或生物化学意义的大型系统。以下两个评论通过示例总结了该软件包的功能。
Molpro 非常容易用于标准应用程序,还包括许多用于专家应用程序的高级选项。它在 UNIX/Linux(2.6.32 以上内核)和 OS-X(Mountain Lion 以上)下运行。Molpro 也可以用作开发平台。
● 生成一般收缩对称化高斯基的积分(spdfghi)。
● 有效核势(ECP)和核极化势(CPP)。
● 闭壳层和开壳层(包括自旋约束和非约束)自洽场方法(RHF,UHF)。
● Kohn-Sham 框架下的密度泛函理论(DFT),支持大量一般梯度近似(GGA)交换泛函和相关泛函。
● 计算量相对于分子尺度增长为线性标度的局域电子相关方法(LMP2, LQCISD(T), LCCSD(T))。
● 可以直接计算激发能的CIS, CC2 和EOM-CCSD 方法。
● 使用直接积分技术(Integral-direct)的Hartree- Fock,DFT,MCSCF 和电子对相关方法(MPn, CCSD, MRCI)。
● 以及一些其他方法等
● 可以计算解析能量梯度的方法有:SCF, DFT, MP2, QCISD(T), LMP2,LQCISD, SA-MCSCF/CASSCF, MR-PT2/CASPT2。密度拟合方法已应用于SCF,DFT和LMP2的梯度计算。
● 闭壳层HF 和(单态,single state)MCSCF/ CASSCF 的解析能量二阶导数(Hessians)。
● 在所有方法中都能计算数值能量梯度和数值能量二阶导数(Hessians)。
● 自动进行几何构型优化,过渡态寻找,反应路径跟踪。
● MCSCF 的解析非绝热耦合(non-adiabatic coupling)矩阵元计算,MRCI 的数值非绝热耦合(non-adiabatic coupling)矩阵元计算。
● 点电荷阵列的能量梯度(为做QM/MM优化)
● 以下方法可以计算多种单电子性质:HF, DFT, MCSCF/CASSCF, MRCI, EOM-CCSD。使用以 下方法计算多种跃迁性质(Transition Properties:)MCSCF/CASSCF, MRCI, EOMCCSD(CASSCF和MRCI也可计算不同的轨道 组成的波函数之间的跃迁)。MCSCF/CASSCF 能计算一些双电子性质(例如角动量算符)。
● CASSCF 和MRCI 的自旋-轨道耦合(Spin-orbit coupling)。
● 使用有效核势方法或使用Breit- Pauli 形式。
● Mulliken 电荷分析和多极矩分布分析。
● 以及一些其他性质计算等。
● 可视化程序:MOLDEN,XMOL等。
● QM/MM程序:Chemshell,QoMMMa,QMPOT等。
● WebMO。
● MRCC程序(Kallay & Gauss)。
● 以XML格式输出CML几何构型和所有结果,基组,轨道,可以在浏览器上看到其中包含的三维几何构型。
● 修复了有关使用 cartesian basis functions 的各种问题,包括起始猜测和 AVAS。密度拟合现在适用于 h 函数的笛卡尔基组。
● DFT grid option orient 的默认值已更改为 1;这使得 KS 能量旋转不变,但可能会稍微改变能量(主要在 microhartree 范围内)。要恢复旧行为,请添加网格选项 orient=1。
● PNO-LCCSD 程序中添加了 一个新的 F12 投影仪选项。现在支持不同的F12价、核价和核-核对双子指数。
● RS2C 期望值计算的内存使用和性能显着提高。添加了NOPROP选项以跳过期望值计算。
● 用于控制网格精度的 DFT 选项已添加到 gmolpro 中。
● gmolpro 添加了用于选择优化方法和坐标的 OPTG 选项。

A bug affecting KS and TDDFT calculations with cartesian basis functions has been fixed.
Perturbation-Adapted Perturbation Theory (PAPT)
The perturbation-adapted zero-order hamiltonian described in J. Chem. Phys. is available for closed-shell systems.
CORE directive
With CORE,MIXED correlation of the 2s and 2p electrons for the second-row elements Al - Ar is now excluded. Global CORE command is now supported. The old behaviour, where these electrons were correlated, can be recovered using CORE,MIXED_OLD .
SCF program
● Quadratic optimization or mixed quadratic/first-order SO-SCI optimization is available for closed-shell and open-shell RHF by using {RHF,SO;…} or {RHF,SO-SCI;…} .
● The CAHF optimization with the quadratic or the SO-SCI optimization is implemented into the CAHF program, and it can be called by {CAHF,SO;…} or {CAHF,SO-SCI;…} .
● A new variant of the SO-SCI optimization is available for the RHF case. It optimizes all AVAS orbitals quadratically and is called by {RHF,SO-SCI-ACTIVE} .
The SO-SCI option significantly improves convergence and is recommended in difficult cases. The cost may even be lower than for standard ROHF or CAHF calculations, since the number of iterations is reduced. Furthermore, the probability of convergence to local minima or saddle points is much reduced.
MCSCF/CASSCF
● The entire export of CI vectors to subsequent Molpro programs has been rewritten. CI vectors can be exported via SAVE,CIREC=record or NATORB,orbrecord,CIREC=record ; more details can be found here. All exported CI vectors are stored in the CSFs basis. Determinants are automatically transformed into the CSFs basis, which also allows an export with state-averaging over different spins. Warning: MULTI based CI records from earlier Molpro versions are not compatible anymore, and the wfu file has to be recalculated with Molpro 2022!
● {MULTI,CASCI;…} can now be simply called by {CASCI;…} , and the input orbitals are not transformed after the CASCI optimization (also for dont,orbital).
● Gradients of the state-averaged energy are now available.
● The program is now more robust against spin-contamination in determinant based calculations.
MRCI, CASPT2
The CI vectors stored in multi using the directive SAVE,CIREC=record can now be used in the MRCI and CASPT2 programs as reference vectors using option CIREC=record .
MCSCF/CASSCF spin-orbit calculations (HLSMAT)
A new much more efficient algorithm for computing the spin-orbit Hamiltonian using MCSCF wavefunctions and ECP spin-orbit operators. The program to compute and diagonalize spin-orbit matrices can generally be called using
HLSMAT,type, record1, record2, record3, …
where record1 , record2 etc are records saved with MULTI or MRCI. The new program is used if type is ECPLS and if no external excitations are present (so either MCSCF/CASSCF wavefunctions or MRCI ones with NOEXC).
PNO-LCCSD
The IBO localization convergence threshold has been tightened from 10-8 to 10-9. The PAO redundancy threshold THRLOC has been tightened from 10-6 to 10-7.
The localisation of core orbitals has been improved; it now uses an AO-based algorithm, which minimises mixings of orbitals of different types (e.g. s, px, py, pz). It has been found that this stabilises the F12(PNO) energy contribution if core orbitals are correlated. The new default for core-orbital localization is loc_method_core=IBO(AO).
The previous behaviour of the PNO program can be recovered by putting before the first PNO command:
local, thrpip=1.d-8, thrloc=1.d-6, loc_method_core=ibo
The corresponding new options are
local, thrpip=1.d-9, thrloc=1.d-7, loc_method_core=ibo(ao)
The options can also be given on the PNO command, lines.
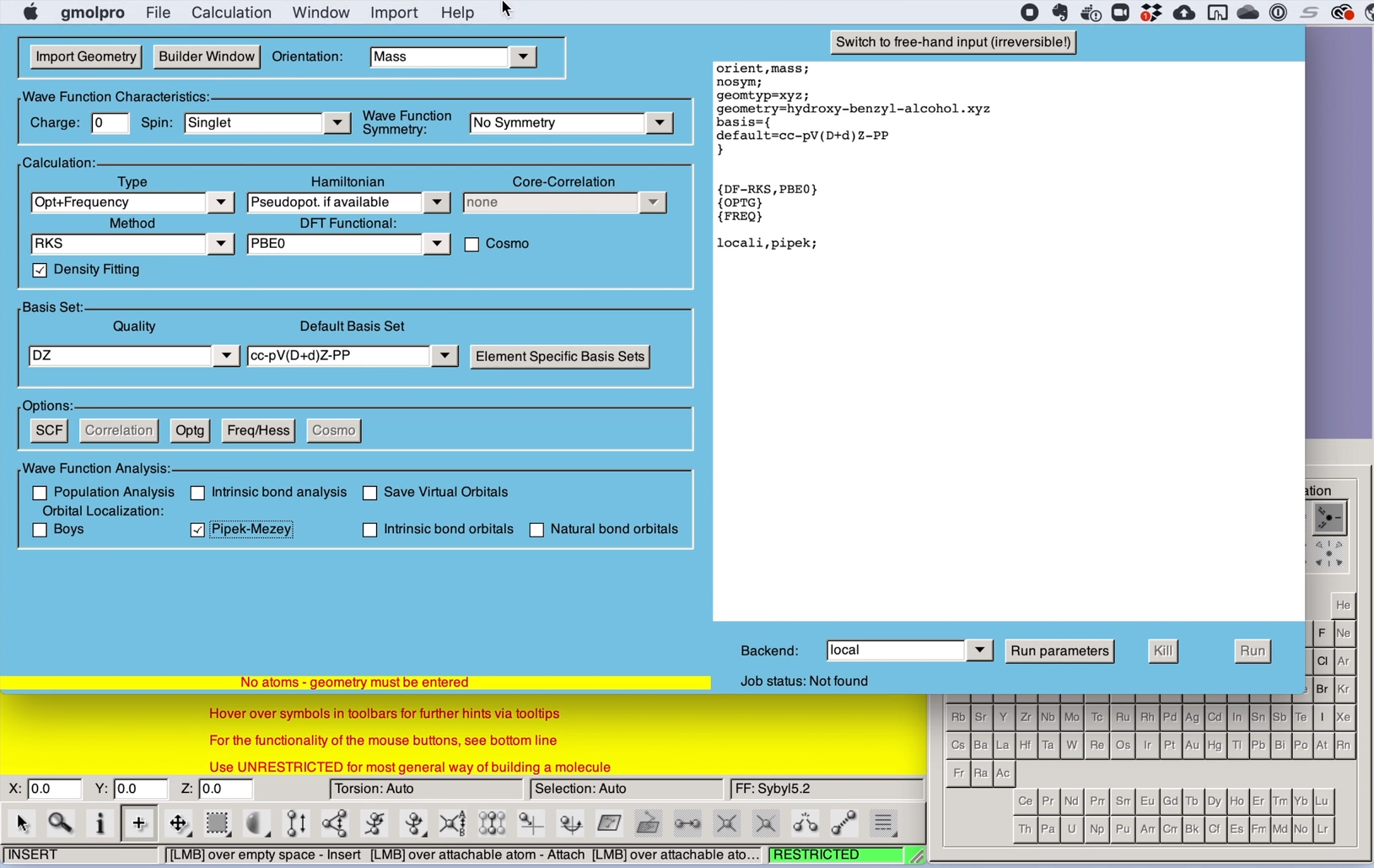
GUI gmolpro
Version 1.4.0 of the graphical user interface gmolpro is compatible with Molpro2022.1 .
Main improvement over earlier versions is the ability to handle output files (xml files) with several optimisations, frequency calculations, or even different molecules within one xml file. Start from the brown window, open the orbital window, and then select and load an orbital set. A corresponding optimisation or vibrational modes may now be displayed.
When creating an input in guided mode, more properties may be selected by buttons.
 北京友万信息科技有限公司,英文全称:Beijing Uone Info&Tech Co.,Ltd ( Uone-Tech )是中国大陆领先的教育和科学软件分销商,已在中国300多所高校建立了可靠的分销渠道。拥有最成功的教学资源和数据管理专家。如需申请软件采购及老版本更新升级请联系我们,咨询热线:010-56548231 ,咨询邮箱:info@uone-tech.cn 感谢您的支持与关注。
北京友万信息科技有限公司,英文全称:Beijing Uone Info&Tech Co.,Ltd ( Uone-Tech )是中国大陆领先的教育和科学软件分销商,已在中国300多所高校建立了可靠的分销渠道。拥有最成功的教学资源和数据管理专家。如需申请软件采购及老版本更新升级请联系我们,咨询热线:010-56548231 ,咨询邮箱:info@uone-tech.cn 感谢您的支持与关注。
地址:北京市昌平区中兴路21号院4号楼5层516 网站备案号:京ICP备16049373号-1]
联系方式:+86-10-56548231
