-

-
-

-
-

-
-

- 010-56548231
-

![]()

![]()

![]()

![]()
![]()
 Q-Chem 6—分子密度泛函计算
Q-Chem 6—分子密度泛函计算
Q-Chem是由遍及全球的多个研究机构共同开发的从头计算量子化学软件包,涉及的科研机构有35家,分布在美国、英国、德国、澳大利亚、中国和中国台湾地区等国家和地区。最早是由诺贝尔化学奖获得者John. A. Pople主导开发的。
Q-Chem软件全面支持HF/DFT以及各种post–HF计算方法,在理论化学、材料科学、生物化学等相关领域的研究和生产中发挥了极大作用。可以研究分子能量和结构、化学反应、分子振动、红外以及拉曼光谱、NMR谱等,还可实现对生物大分子的QM/MM 计算。从1999年第一个版本Q-Chem1.2发布至今,Q-Chem软件已经开发了15年,成为一个功能十分强大的量子化学计算程序。
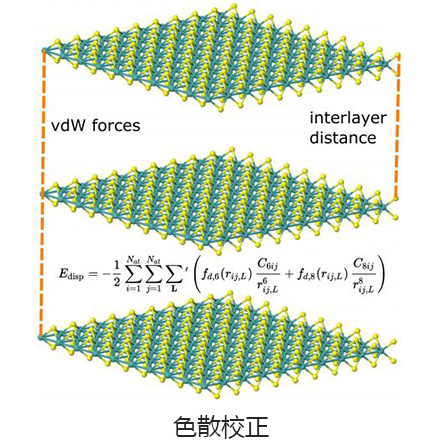
Q-Chem支持LDA,GGA和meta-GGA功能,以及GGA和meta-GGA的混合,范围分隔混合和双混合版本。可以通过与时间有关的DFT评估基态和激发态的单点能量,几何形状优化,振动频率计算以及许多其他属性。
Q-Chem提供了用于处理电子相关效应的最新工具,例如Møller-Plesset摄动理论和耦合簇理论。对于具有强相关性的系统,Q-Chem提供特殊处理,包括CASSCF,耦合簇价键理论,选定的CI,RAS-CI,自旋翻转和变分2-RDM方法。
Q-Chem提供了多种方法来研究电子激发态:CIS,TD-DFT,NOCI,EOM-CC和ADC。这些方法的特殊风味涵盖了许多类型的电子结构,从而可以模拟光谱特征,电荷和能量转移以及非绝热动力学。

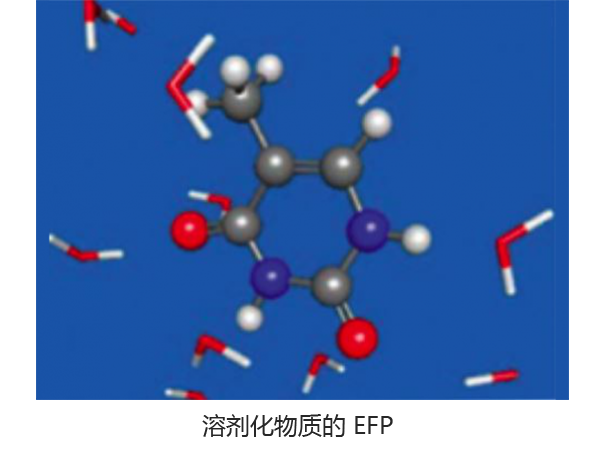
Q-Chem 软件包为溶剂化系统建模提供了多种解决方案,从隐式溶剂模型(如 SM8、COSMO 和 C-PCM)到有效片段电位法(可用于捕获显式溶剂效应)。此外,Q-Chem 包括几种不同的嵌入方法,包括 QM/MM 和密度嵌入,以及与 CHARMM 和 GROMACS 的接口。
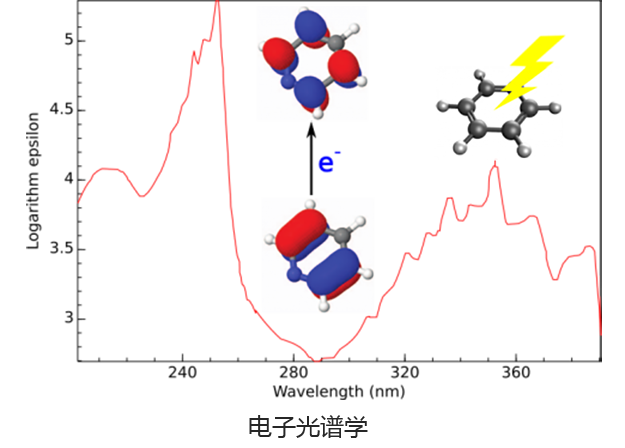
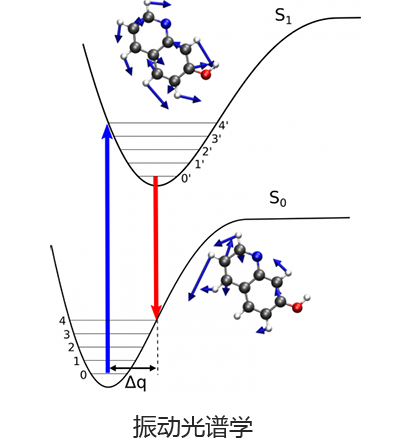

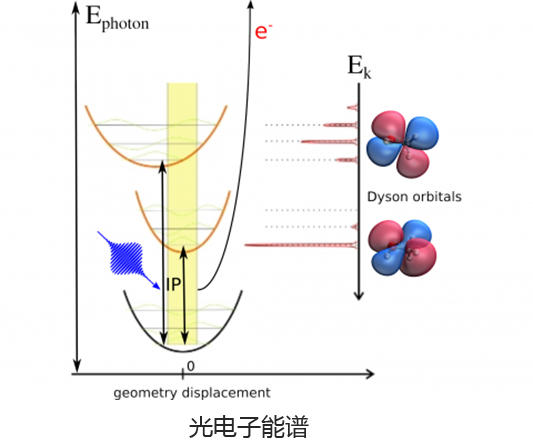
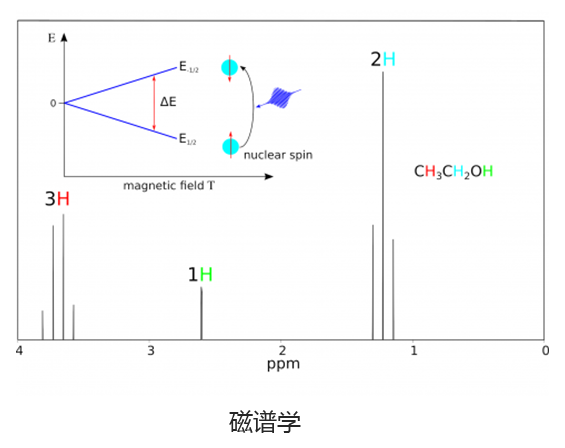
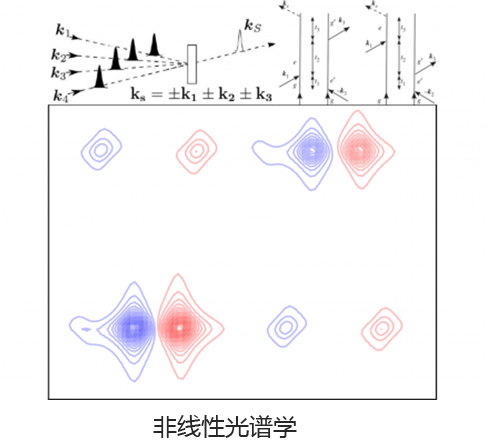

Q-Chem 提供了多种工具来模拟不同类型的光谱。我们的能力包括红外和拉曼光谱、紫外可见光谱、X 射线光谱、光电子光谱、核磁共振光谱和非线性光谱(例如双光子吸收)。可以使用许多不同层次的理论来研究光谱特征,从 TDDFT 到 EOM-CC 和 ADC 方法。
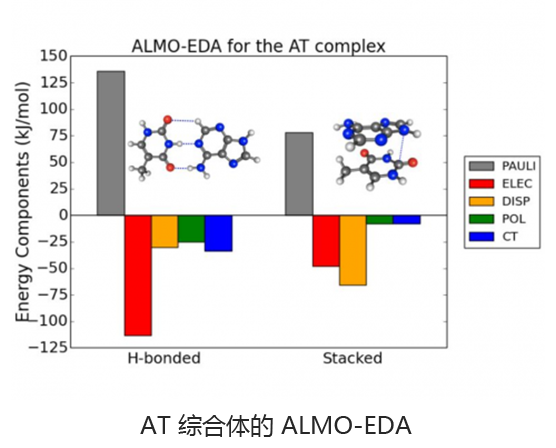
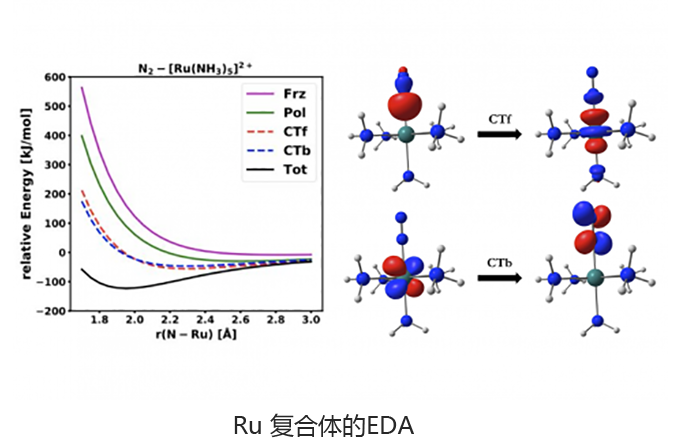
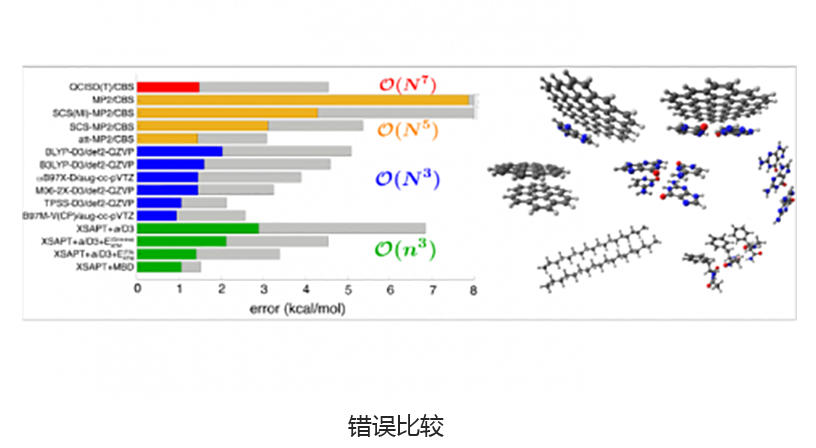
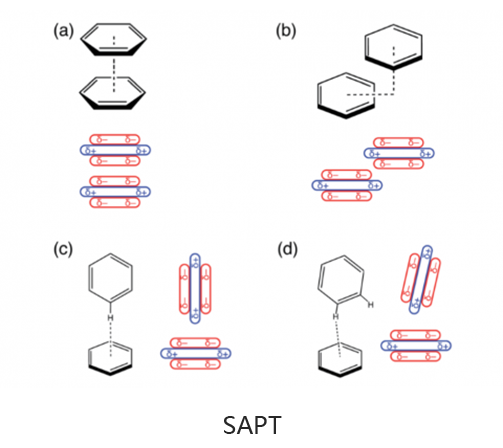

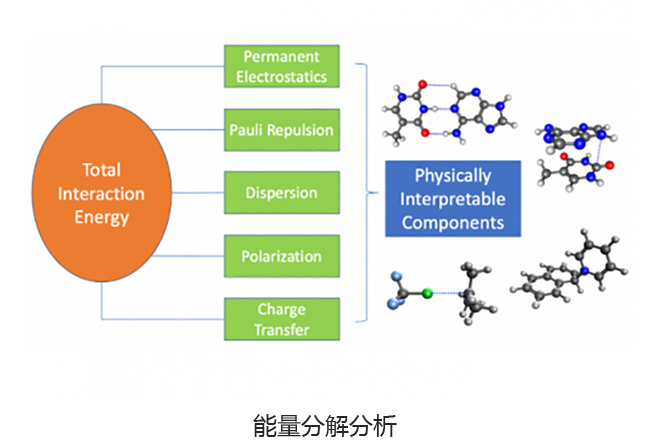
基于绝对局部分子轨道的能量分解分析可将总相互作用能分解为有意义的物理术语,从而洞悉分子间和键合相互作用的性质。适应对称的扰动理论(SAPT)及其扩展的多体形式(XSAPT)也可用于计算和分析分子间的相互作用。
Q-Chem 提供几何优化、势能表面扫描、过渡态搜索和本征反应坐标跟踪的方法,使其成为化学反应性、热化学和化学动力学研究的理想选择。

Q-Chem 可以执行 从头算分子动力学 (AIMD),包括 NVE 和 NVT 热采样,以及准经典分子动力学 (QMD)。这些方法可用于产生振动光谱和从头算路径积分。我们还实施了 Tully 的最少开关表面跳跃 (FSSH) 方法,以有效处理非绝热系统。
无需购买额外的可视化软件。
IQmol是Q-Chem的配套可视化软件,可以免费获取。Q-Chem也支持第三方可视化软件,如Avogadro, WebMO, Jmol等;
无需购买额外的并行许可即可进行跨节点并行计算。
程序的代码经过专门的优化,保证更快的计算速度;
支持多种方法分析势能面。
如冻结带方法、从头算分子动力学、路径积分蒙特卡洛方法及偏Hessian分析;
可进行能量组成分析。如可以把能量分解成库伦项、交换项、极化项及电荷转移项;
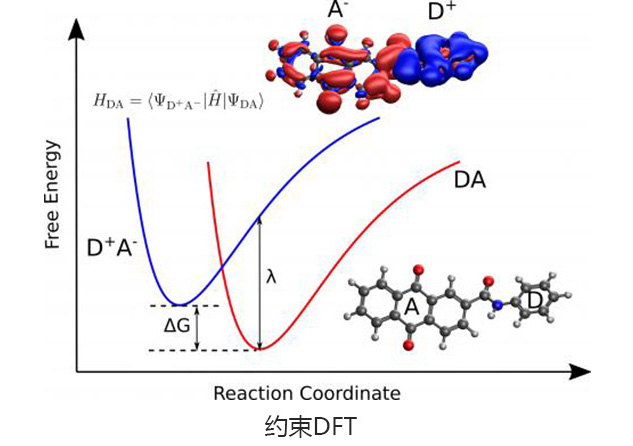
可研究电子转移耦合及激发能。支持的方法有限制性DFT、直接耦合及分子片段电荷;
拥有独特的基态泛函。
如色散校正泛函XDM、DF-04、XYGJ-OS、WB97X-2等;
快速和更加精确的MP2方法。如SOS-MP2、双基组MP2及局域MP2方法;
支持激发态研究(包括开壳层分子)。如SOS-CIS(D)、TDDFT、ADC等;
Q-Chem 提供优秀的个人技术支持服务,并会依据客户的要求开发所需的功能。
势能面扫描
Q-Chem能够高度精确的给出分子在不同构型下的能量,帮助我们找到分子局部的能量极小值以获得稳定的分子结构。还可以根据分子的构象进行势能面扫描。
分子间相互作用
Q-Chem采用Hartree-Fork,基于各种泛函的DFT方法和高精度的电子相关方法,多体微扰的MPn与Coupled-cluster理论来计算分子间的相互作用,并可以非常便捷的进行BSSE矫正。此外,Q-Chem还可以基于ALMO方法将分子之间的相互作用能进行分解。
过渡态搜索和反应路径分析
Q-Chem是业界公认的研究化学反应的有力工具。Q-Chem支持多种过渡态搜索的方法,并可进一步对反应路径进行分析和优化。得益于Q-Chem软件的高效率,研究者可以快速方便的研究各种化学反应相关问题。
激发态和光谱计算
除了研究基态的化学性质外,Q-Chem还支持激发态结构的能量计算和优化,新版本的Q-Chem还加入了M11等最新的泛函并支持EOM-CCSD方法。
Q-Chem可以计算各种光谱性质,包括:IR, Raman, 紫外-可见,VCD,ECD等。
线性标度的量化计算
Q-Chem支持多种线性标度的量子化学计算方法,包括:傅立叶变换库仑方法,线性标度HF交换方法,基于格点的线性标度积分等。
QM/MM 计算
Q-Chem支持ONIOM计算并自带了与分子动力学模拟软件CHARMM的程序接口。可以非常方便的进行QM/MM计算。
基态自洽场方法
1. Hartree-Fock方法
限制性,非限制性,和限制性开壳层形式
用于结构优化的解析一阶导数
用于谐振频率分析的解析二阶导数
2. 密度泛函理论
局域泛函和梯度校正泛函。交换泛函:Slater,Becke'88,Perdew'91,Gill'96,Gilbert-Gill'99,Handy-Cohen OPTX。关联泛函:VWN5,Lee-Yang-Parr,Perdew-Zunger'81,Perdew'86,Wigner,Perdew'91。EDF1交换-关联泛函。用户定义的交换-关联泛函。
HF-DFT杂化泛函:B3LYP,B3PW91,B3LYP5,用户定义的杂化泛函。
基于数值格点的数值积分方案:SG-0标准网格,SG-1标准网格,Lebedev和Gauss-Legendre角向积分方案
用于结构优化的解析一阶导数
用于谐振频率分析的解析二阶导数
3. 线性标度方法
傅立叶变换库仑方法
连续快速多极方法
线性标度HF交换方法
基于格点的线性标度积分,用于交换-关联泛函求值
线性标度NMR化学位移
4. AOINTS包用于双电子积分
结合了高性能积分技术的最新进展;COLD PRISM;J-矩阵引擎。
5. SCF改进
in-core和直接SCF的最优混合
DIIS
初始猜测方案:重叠球平均原子密度,广义Wolfsberg-Helmholtz,从小基组投影,芯哈密顿量的猜测
SCF波函的稳定性分析
最大重叠方法
Fock矩阵的直接最小化
极化原子轨道对分子优化的最小基
基于波函的电子关联处理
1. Møller-Plesset微扰理论
限制性,非限制性,和限制性开壳层形式
直接和半直接方法计算能量
半直接方法的解析梯度,用于限制性和非限制性形式
在MP3,MP4和MP4SDQ方法的解析梯度计算中处理冻芯轨道
2. 局域MP2方法
根据物理图象截断完全MP2的能量表达式,从而减少计算量
减少计算量相对于分子尺寸的标度,近似为两倍,却不明显丢失精度。
应用外推PAO用于局域校正
可以使用分子中的双原子和分子中的三原子技术
3. RI-MP2
比MP2和局域MP2快十倍
4. 耦合簇方法
CCSD:能量,以及作为能量有限差分的梯度
EOM-XX-CCSD;XX = EE, EA, IP, SF,能够灵活处理自由基,键的断裂,以及对称破缺问题
耦合簇能量的非迭代校正:三级校正CCSD(T),三级和四级校正CCSD(2)
广泛应用分子点群对称性,以改善效率
二次双激发耦合簇
QCISD,QCISD(T)和QCISD(2)用于能量
DIIS用于收敛加速
冻芯近似,用于增加可处理体系的尺寸
5. 优化轨道的耦合簇方法
优化轨道的双激发耦合簇(OD):可避免人为的对称破缺问题;优化平均场参考轨道使能量最小;Brueckner耦合簇;OD,OD(T),和OD(2)的能量及梯度
优化价轨道的耦合簇方法(VOD):传统CASSCF方法的耦合簇近似;在价活性空间利用截断的OD波函;比CASSCF有更少的磁盘空间需求和更小的体系标度,可处理较大体系;VOD,VOD(T),VQCCD,和VOD(2)的能量及梯度
激发态方法
1. 支持的计算类型
垂直激发吸收谱
通过激发态能量的有限差分,进行激发态的结构优化
UCIS和RCIS进行激发态的振动分析
自旋反转DFT
2. CIS方法
从Hartree-Fock基态波函计算激发态:获得定性的单电子激发态;结构和频率与基态Hartree-Fock结果有可比性
高效的直接算法用于计算闭壳层和开壳层体系的能量、解析梯度和二阶导数
XCIS用于二重和四重态计算
双激发微扰校正CIS(D),可使CIS误差减少两倍或更多,接近于MP2
3. TDDFT
从Kohn-Sham基态波函计算激发态能量
对于低位价激发态,TDDFT比CIS有相当大的改善,但只有相近的计算量
提供激发态中关联效应的内在图像
自由基的低位价激发态,比CIS有相当大的改善
自旋反转密度泛函理论(SFDFT):把TDDFT推广到低位价激发态之外;可用于键断裂的过程,以及自由基和双自由基体系。
4. 基于耦合簇的激发态方法
EOM-CCSD
自旋反转激发态方法:改善了双、三自由基体系的处理;结合单行列式波函处理键断裂问题;可用于OD和CCSD理论级别。
OOD方法:与CCSD激发态方法有几乎相同的数值性能;比TDDFT精度更高,但计算量更昂贵。
EOM-VOOD方法:类似于EOM-CCSD,但使用VOOD方案。
激发态特性计算:跃迁偶极矩和结构。
5. 分解分析
显示电子跃迁的工具,用于把电子跃迁分类为价跃迁、Rydberg跃迁,混合跃迁,或电荷转换。
1. 自动结构优化和过渡态优化
使用Jon Baker博士的OPTIMIZE程序包,用约化内坐标保证迅速收敛,避免初始力常数矩阵
具有一般约束的结构优化:可施加于键角,二面(扭转)角,或平面外的弯曲;直角坐标中冻结原子;约束不一定要加在初始结构上
优化使用笛卡尔,Z-矩阵或离域内坐标
本征矢跟踪算法,用于过渡态和最小化
GDIIS算法用于最小化:使到平衡结构的收敛获得极大加速
内反应坐标跟踪:沿着反应路径的连续平衡结构和过渡态
2. 振动光谱
自动调用解析和数值二阶导数
红外和拉曼强度
输出标准的统计热力学信息
同位素替换,用于与实验进行比较
非谐性校正
3. NMR屏蔽张量
4. 自然键轨道分析
使用NBO 5.0
5. Stewart原子
从分子密度重新获得原子特性
Q-Chem用恒等解方法计算这些值
6. 动量密度
7. Intracules
独特的双电子函数,提供分子中库仑能和交换能关于位置和动量的最详尽信息
8. 分子中的原子
利用免费的AIMPAC进行AIM分析
9. 溶解模型
简单的Onsager反应场模型
Langevin偶极模型
SS(V)PE:一种新的电解质连续模型
10.基于Dirac-Fock理论的相对论能量校正
11.对角绝热校正
计算Born-Oppenheimer对角修正,研究核与电子运动绝热距离的分解
基组
1. 高斯基组
2. 赝势基组
3. 用户定义的基组和赝势
4. 基组重叠误差(BSSE)校正
QM/MM
1. 到CHARMM的接口
2. ONIUM
其他
1. Q-Chem的功能已经完全整合到Spartan程序中,在图形用户界面下计算更容易
Q-chem 6 新版本特点
——Q-Chem 与外部工具的下一代接口(生成 HDF5 格式的存档文件)
——用于基态和激发态计算的新几何优化器
——使用VV10 泛函的解析频率计算和轨道 Hessians (Jiashu Liang)
——核电子轨道 (NEO) 系列方法的发展,包括NEO-CCSD和NEO-TDDFT 的分析梯度和 Hessians(Zhen (Coraline) Tao, Patrick E. Schneider, Fabijan Pavosevic, Sharon Hammes-Schiffer)
——CCSD 旋光度评估 (Josefine Andersen, Kaushik Nanda)
Q-Chem 6.0.1 发布
2022 年 8 月 24 日
更改默认行为
恢复了对 CHARMM (John Herbert) 使用的 IGDESP 的支持
实现了内存高效的 GOSTSHYP 算法 (Felix Zeller)
使用与规范无关的原子轨道 (GIAO) 为 HF 和 MP2 启用有限场化学屏蔽和磁化率计算(Jonathan Wong、Brad Ganoe、Tim Neudecker、Adam Rettig、Xiao Liu、Joonho Lee)
已解决的问题:
用于 libopt3 中优化的模塑输出
使用 PCM 的 libopt3 Hessian 计算
将 MO 系数和能量写入 qarchive 文件
清理拓扑检查打印
在几何优化期间固定打印步长的 RMS
分子动力学、非绝热动力学、嵌入和溶剂化
使用 RAS-SF (Bushra Alam, Hanjie Jiang, John Herbert, Paul Zimmerman) 实现了特定于状态的 PCM
碎片和能量分解分析
解决了基于投影的嵌入计算问题,其中冻结(环境)占据的轨道不是基于能量排序的(Yuezhi Mao)
各种各样的
在 DFT SOC 计算中添加了用于识别作业进度的注释(Saikiran Kotaru)
添加了关于某些优化作业没有分析 Hessians 的警告
添加打印以区分 CPCM1 和 CPCM2
Q-Chem 6.0.0 发布
2022 年 7 月 3 日
更改默认行为
• 将默认积分阈值($rem 变量 THRESH)收紧至 SCF_CONVERGENCE + 4,并为 DIIS 和 GDM 使用相同的阈值
• 将 VV10 功能的 FD_MAT_VEC_PROD 的默认值设置为 FALSE (Yuezhi Mao)
• 关闭网格上静电势的自动评估 (Felix Plasser)
• 为电场中的二阶能量导数设置有限差分为默认值 (Yuezhi Mao)
一般功能和改进
• 下一代 Q-Chem 接口与外部工具(生成 HDF5 格式的存档文件)
• 实施了核电子轨道 CCSD (NEO-CCSD) 方法 (Fabijan Pavosevic, Sharon Hammes-Schiffer)
• 实施 NEO-TDDFT 分析梯度和 Hessian (Zhen (Coraline) Tao, Patrick E. Schneider, Sharon Hammes-Schiffer)
• 通过自旋输入部分在 NMR J 耦合计算 (JOBTYPE = ISSC) 中启用原子子集选择
• 在固定原子的几何优化中禁用最速下降
• 在新优化器中添加了离域自然内部坐标优化
• 为有限差分优化中的每个步骤更新了 MOLDEN 文件中的几何图形 (John Herbert)
• JK 和 MP2 的稳定密度拟合
• 将新优化器设置为无约束优化的默认值 (GEOM_OPT_DRIVER=2022)
• 添加了卡尔斯鲁厄基组的最小增广和重增广版本 (John Herbert)
• 删除了 MPI 支持
• 已解决的问题:
▸ 基于分子输入顺序的不正确赫什菲尔德收费 (Abdulrahman Aldossary)
▸ SOC 计算中的非数字 (NAN) 错误
▸ 福克投影(基础2)计算中缺少核排斥能
▸ 删除了对可包含在随机搜索和盆地跳跃中的原子数 (MAX_ATOM) 的限制
▸ 格式化检查点文件中本地化 MO 的排序 (Abdulrahman Aldossary)
▸ def2-SVPD 基组缺少 ECP
▸ 无法计算具有线性相关基组的 NMR 属性
▸ 解析超过 100k 行的输入文件
▸ C3点群字符表
密度泛函理论与自洽场
• 加速 SCF 算法 ADIIS 的收敛并添加新的组合算法选项 ADIIS_DIIS。(毛月之)
• 使用 gen_scfman(Brad Ganoe、Tim Neudecker、Joonho Lee、Adam Rettig、Jonathan Wong)在 SCF 计算中启用与规范无关的原子轨道 (GIAO)
• 如果使用内置的范围分隔功能,则禁用用户设置系数(通过 HFK_LR_COEF/HFK_SR_COEF)
• 为 VV10 泛函实现了频率计算和解析 Hessian (Jiashu Liang)
• 使用复杂的基函数实现基于投影的嵌入 (Valentina Parravicini, Thomas Jagau)
• 通过 GUI = 2 (Yuezhi Mao) 启用在 CIS/TDDFT 计算中使用冻结的占用/虚拟轨道生成格式化的检查点文件
• 为新的情节部分启用 STATE_ANALYSIS (PLOT=1) (Yuezhi Mao)
• 基于 SCF 收敛阈值 (SCF_CONVERGENCE) 而不是场幅度 (John Herbert) 对 TDKS Fock 矩阵执行一致性检查
• 添加了新的能量密度泛函:revSCAN、regSCAN、r++SCAN、r2SCAN、r4SCAN、TASK、mTASK、regTM、rregTM、revTM
• 使用 TDDFT(受限和非受限)和自旋翻转 TDDFT (SF-TDDFT) 计算自旋轨道耦合 (SOC)(1 电子和 2 电子平均场)
• 为自交互校正(Marc Coons、Bhaskar Rana、John Herbert)实施密度校正 DFT (DC-DFT) 的分析梯度
• 已解决的问题:
▸ 使用 gen_scfman (Yuezhi Mao) 计算分数电子 SCF 的结果不正确
▸ 使用大量 OpenMP 线程挂起 qints (use_libqints = true) 作业
▸ ADIIS 的非变分初始 SCF 猜测(Yuezhi Mao)
▸ TDDFT/TDA 计算中的内存估计不正确
▸ TDA激发态频率工作的崩溃
▸ 固定原子几何优化的崩溃
▸ 使用具有 g 或更高角动量的基函数进行频率计算
▸ TDDFT 自旋轨道耦合计算的符号错误 (Nicole Bellonzi)
▸ 基于投影的嵌入计算崩溃(Yuezhi Mao)
▸ 使用非波普尔基组的 RPA TDDFT 频率结果不正确
▸ 使用 meta-GGA 泛函进行 NMR 计算的内存分配不足
▸ 使用具有较大基组的混合泛函在 DC-DFT 计算中的错误结果 (Marc Coons, Bhaskar Rana, John Herbert)
▸ 使用 CIS/TDDFT 的激发态势能表面扫描的碰撞 (John Herbert)
相关方法
• 在速度和混合仪表中实施 EOM 振荡器强度 (Josefine Andersen, Sonia Coriani)
• 实施 CCSD 旋光评估 (Josefine Andersen, Kaushik Nanda)
• 在 rasman2 (Chou-Hsun (Jeff) Yang, Aaditya Manjanath, Chao-Ping (Cherri) Hsu) 中实施了碎片电荷差异 (FCD) 方案
• 实施复值 CC2、RI-CC2 和 RI-CCSD(Cansu Utku、Garrette Paran、Thomas Jagau)
• 在 AIMD 计算中实施复吸收势 (CAP) 方法 (Jerryman A. Gyamfi, Thomas Jagau)
• 使用密度拟合基组实施 v2RDM-CASSCF-PDFT 方法(Mohammad Mostafanejad、Run Li、A. Eugene DePrince III)
• 已解决的问题:
▸ 使用 RAS-CI 方法计算 SOC 输出中的格式错误 (Abel Carreras, David Casanova)
分子动力学、非绝热动力学、嵌入和溶剂化
• 为泊松方程求解器 (PEqS) 启用用户定义的介电常数网格 (Suranjan Paul)
• 改进的 PCM 打印 (John Herbert)
• 实施 CIS 和 TDDFT 波函数重叠,包括 (A)FSSH 的自旋翻转变体(Theta Chen、Junhan Chen、Zuxin Jin、Vishikh Athavale、Vale Cofer-Shabica、Joe Subotnik)
• 已解决的问题:
▸ QM/MM 优化不读取以前的 MO 作为下一个周期的猜测
碎片和能量分解分析
• 在 QM/EFP 计算中实施成对碎片激发能量分解分析 (EDA) (Lyudmila Slipchenko)
• 将 XSAPT 计算的基函数的最大角动量增加到 5
• 实施了基于 SPADE 和 ALMO 的电场计算分区方案(Yuezhi Mao)
• 实施了新的 MP2 EDA 方案并为 DFT EDA 添加了非微扰偏振分析(Kevin Ikeda,Hengyuan Shen)
• 启用 ALMO-CIS/TDA 计算,激发振幅位于一个片段上 (Yuezhi Mao)
• 启用 ALMO-CIS/TDA 计算,激发从一个碎片的占据轨道到系统中的所有虚拟 (Yuezhi Mao)
• 启用 ALMO-CIS/TDA 计算,激发从一个片段的占据轨道到另一个片段的虚拟轨道 (Yuezhi Mao)
• 在 ALMO-CIS/TDA 计算中启用用户定义的占用虚拟对 (Yuezhi Mao)
• 解决了 ALMO-CIS 和激发态 ALMO-EDA 计算的杂项问题 (Yuezhi Mao)
各种各样的
• 使用 **SCF_PRINT = 3** 打印的轨道动能
• 在外部文件中启用 EXTERNAL_CHARGES 规范 (Vale Cofer-Shabica, Joseph Subotnik)
• 为多体色散计算添加了参数检查 (John Herbert)
• wB97M2 和 XYG 系列能量泛函的恢复有限差分
• 恢复 SA-SF-RPA 的有限差分横幅
Q-Chem runs on a wide variety of computer systems, ranging from Intel and AMD microprocessor-based PCs and workstations, to high-performance server nodes used in clusters and supercomputers. Q-Chem supports the Linux, Mac, and Windows operating systems.
Memory
Q-Chem, Inc. has endeavored to minimize memory requirements and maximize the efficiency of memory usage. Still, the larger the structure or the higher the level of theory, the more memory is needed. Although Q-Chem can be run successfully in very small-memory environments, this is seldom an issue nowadays and we recommend 2 GB per CPU core as a minimum. Q-Chem also offers the ability for user control of important, memory-intensive aspects of the program. In general, the more memory your system has, the larger the calculation you will be able to perform.
Q-Chem uses two types of memory: a chunk of static memory that is used by multiple data sets and managed by the code, and dynamic memory which is allocated using system calls. The size of the static memory is specified by the user through the $rem variable MEM_STATIC and has a default value of 192 MB.
Disk
The Q-Chem executables, shell scripts, auxiliary files, samples and documentation require about 1.4GB of disk space, depending on the platform. The default Q-Chem output, which is printed to the designated output file, is usually only a few kilobytes.
 北京友万信息科技有限公司,英文全称:Beijing Uone Info&Tech Co.,Ltd ( Uone-Tech )是中国大陆领先的教育和科学软件分销商,已在中国300多所高校建立了可靠的分销渠道。拥有最成功的教学资源和数据管理专家。如需申请软件采购及老版本更新升级请联系我们,咨询热线:010-56548231 ,咨询邮箱:info@uone-tech.cn 感谢您的支持与关注。
北京友万信息科技有限公司,英文全称:Beijing Uone Info&Tech Co.,Ltd ( Uone-Tech )是中国大陆领先的教育和科学软件分销商,已在中国300多所高校建立了可靠的分销渠道。拥有最成功的教学资源和数据管理专家。如需申请软件采购及老版本更新升级请联系我们,咨询热线:010-56548231 ,咨询邮箱:info@uone-tech.cn 感谢您的支持与关注。
地址:北京市昌平区中兴路21号院4号楼5层516 网站备案号:京ICP备16049373号-1]
联系方式:+86-10-56548231
